](https://deep-paper.org/en/paper/13328_from_mechanistic_interpr-1780/images/cover.png)
人工智能与生物学的交叉领域在过去十年中产生了一些最引人注目的科学突破。像 AlphaFold 这样的工具已经解决了蛋白质结构预测问题,而蛋白质语言模型 (pLMs) 现在能够以惊人的准确率生成新蛋白质或预测其功能。
然而,这里有个问题。虽然这些模型非常有用,但它们在很大程度上是作为“黑盒”运行的。我们向模型输入一串氨基酸序列,它输出一个结构或功能预测。但是,它是如何做到的?模型是真的“理解”生物物理学,还是仅仅在记忆统计相关性?
如果我们能窥探这些模型的内部,我们不仅能更好地理解人工智能,还可能发现新的生物学知识。这一概念是一篇名为*《从机械可解释性到机制生物学》(From Mechanistic Interpretability to Mechanistic Biology)* 的新论文背后的驱动力,该论文将一种称为稀疏自编码器 (Sparse Autoencoders, SAEs) 的技术应用于 ESM-2 蛋白质语言模型。
本文将介绍研究人员的方法论、他们关于 AI 如何表征蛋白质“概念”的发现,以及这项技术如何使我们不仅能利用 AI 进行预测,还能将其用于生物学发现。
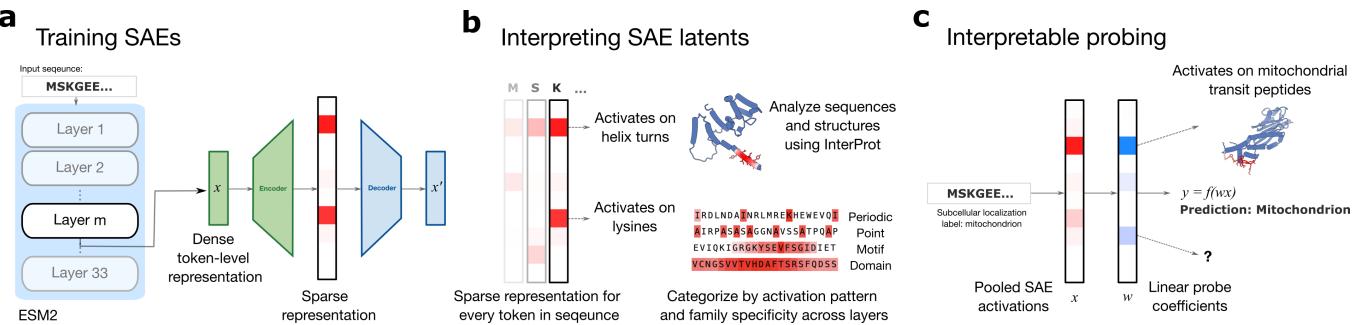
1. 蛋白质模型中的可解释性鸿沟
要理解这项工作的意义,我们必须首先看看蛋白质语言模型 (pLMs) 的现状。像 ESM-2 这样的模型是在数百万个蛋白质序列上通过“掩码词元预测”任务训练出来的。就像 ChatGPT 学习预测句子中的下一个单词一样,ESM-2 学习预测序列中缺失的氨基酸。
在最小化预测误差的过程中,模型学习到了能够捕捉蛋白质结构和功能深刻见解的内部表征 (数字向量) 。生物学家将这些表征用于下游任务,例如预测蛋白质是否具有热稳定性或其在细胞中的位置。
然而,这些内部表征是“稠密”且“多义”的。模型中的单个神经元可能会针对多个不相关的概念被激活——也许它既对疏水性螺旋有反应,又对特定的结合位点有反应。这种纠缠使得我们几乎不可能看着某个特定的神经元说: “这个神经元代表一个 α-螺旋。”
这就是机械可解释性 (Mechanistic Interpretability) 的用武之地。这是一个致力于逆向工程神经网络的领域。最近,大型语言模型 (LLMs) 的研究人员成功地使用稀疏自编码器 (SAEs) 将这些稠密的表征分解为“稀疏”且可解释的特征。这篇论文的作者提出了一个问题: 我们能为蛋白质语言做同样的事情吗?
2. 核心方法: 稀疏自编码器 (SAEs)
SAE 的目标是获取 pLM 混乱、纠缠的激活值,并将它们映射到一个更大、更清晰的特征字典中。
架构
研究人员在 ESM-2 模型 (具体为 6.5 亿参数版本) 不同层的“残差流” (主要信息高速公路) 上训练了 SAE。
SAE 架构主要由两部分组成: 编码器 (Encoder) 和 解码器 (Decoder) 。
编码器接收来自 ESM-2 模型的激活向量 \(x\),并将其投影到一个维数高得多的空间中。这里的关键组件是 “TopK” 激活函数。
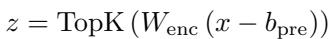
在这个公式中:
- \(x\) 是来自 ESM-2 的输入。
- \(W_{enc}\) 是学习到的权重矩阵。
- TopK 是稀疏性执行器。它强制所有激活值为零,除了最高的 \(k\) 个值。
这种约束迫使模型只选择其字典中最相关的“词”来描述输入的蛋白质。通过强制表征变得稀疏,模型被迫学习独特、有意义的特征,而不是分布式、混乱的特征。
然后, 解码器尝试从这个稀疏表征 (\(z\)) 中重构原始输入。
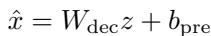
模型致力于最小化重构误差 (\(x\) 和 \(\hat{x}\) 之间的差异) 。如果 SAE 能够仅使用极少数活跃的潜在维度准确重构 ESM-2 的活动,这就意味着这些维度代表了 ESM-2 用于理解蛋白质的基本构建块 (或特征) 。
3. 可视化隐藏的蛋白质语言
训练完 SAE 后,研究人员最终得到了数百万个“潜在特征” (latents)。接下来的挑战是解释它们。与可以阅读输出的英文文本不同,蛋白质序列是晦涩难懂的字母串。
为了解决这个问题,作者开发了 InterProt , 这是一个可视化工具。通过分析哪些序列激活了特定的 SAE 潜在特征,他们可以将这些数学特征映射到生物学结构上。
结果令人震惊。SAE 成功地将模型的知识分解为可识别的生物学概念。
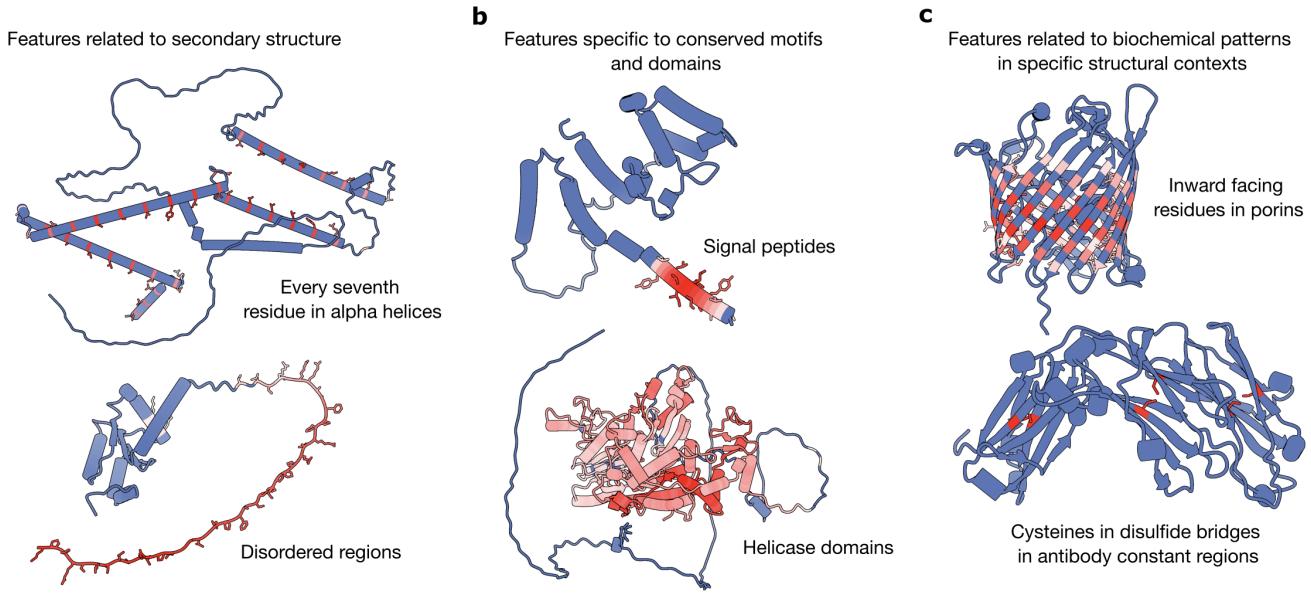
如上图 2 所示,SAE 发现了:
- 二级结构 (图 a): 专门为 α-螺旋或无序区域点亮的特征。
- 模体和结构域 (图 b): 识别信号肽 (指导蛋白质运输) 或特定功能结构域 (如解旋酶) 的特征。
- 生物化学语境 (图 c): 检测特定化学相互作用的特征,例如参与二硫键 (半胱氨酸-半胱氨酸键) 的残基或孔道中朝内的残基。
人类评估
为了确保他们不只是在“挑三拣四”地展示好例子,研究人员进行了一项盲测研究。人类专家对来自 SAE 的特征和来自 ESM 模型的原始神经元进行了评分。
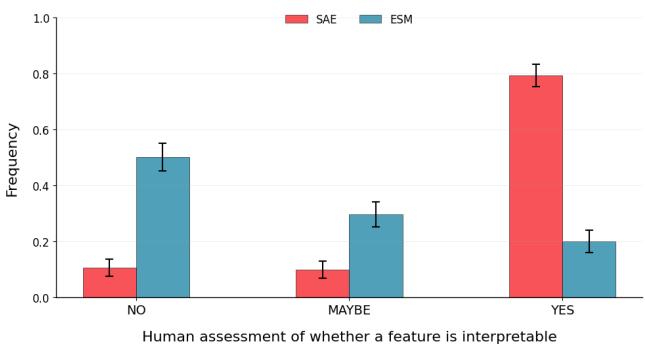
差异是巨大的。如图 4 所示,大约 80% 的 SAE 潜在特征被评为可解释 (“Yes”) ,而原始 ESM 神经元的比例非常低。这证实了 SAE 有效地解开了在基础模型中混合在一起的生物学概念。
4. 蛋白质家族与层级动态
这项研究最引人入胜的发现之一是家族特异性特征 (Family-Specific Features) 的概念。
研究人员发现,许多 SAE 潜在特征并非通用的物理检测器 (如“α-螺旋”) 。相反,它们对进化家族具有高度特异性。一个潜在特征可能会寻找一个螺旋,但仅当它出现在 β-内酰胺酶中时才会被激活。
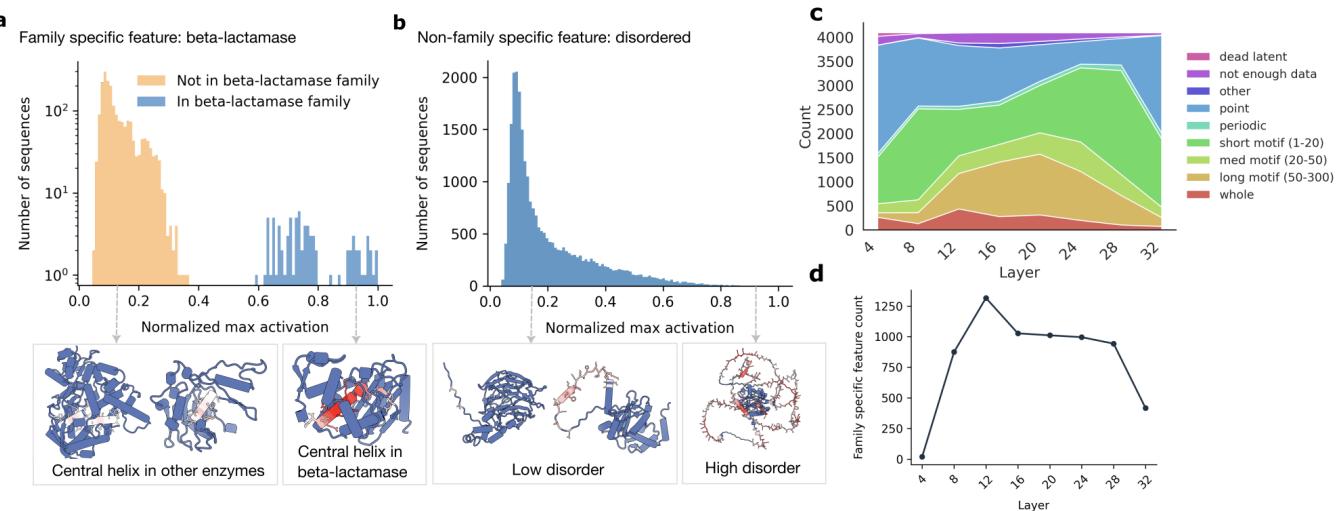
在图 3a 中,我们看到一个潜在特征对 β-内酰胺酶的中心螺旋有强烈激活,但忽略了其他蛋白质中的相似螺旋。将其与图 3b 进行对比,图 3b 显示了一个“无序”特征,它在许多不同类型的蛋白质中广泛激活。
层级解剖
这些特征的分布取决于你在模型中的观察位置。
- 浅层: 倾向于关注局部氨基酸模式和较短的模体。
- 中层: 这是家族特异性达到顶峰的地方 (上图中的图 3d) 。模型似乎在这些层中按进化历史对蛋白质进行分组。
- 深层: 特征变得更加抽象或专门用于最终的预测任务。
激活模式分类
为了系统地分析数百万个特征,作者建立了一个基于特征如何在序列上点亮的分类方案。
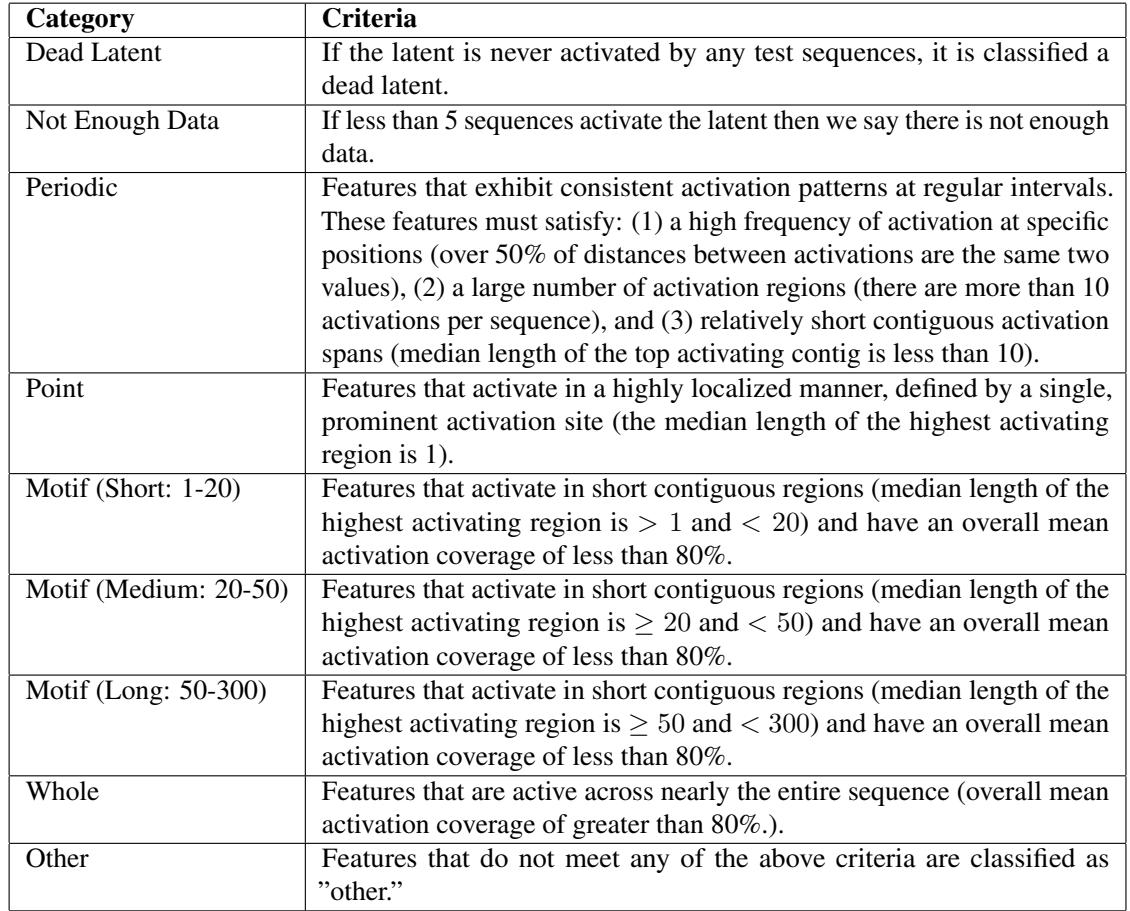
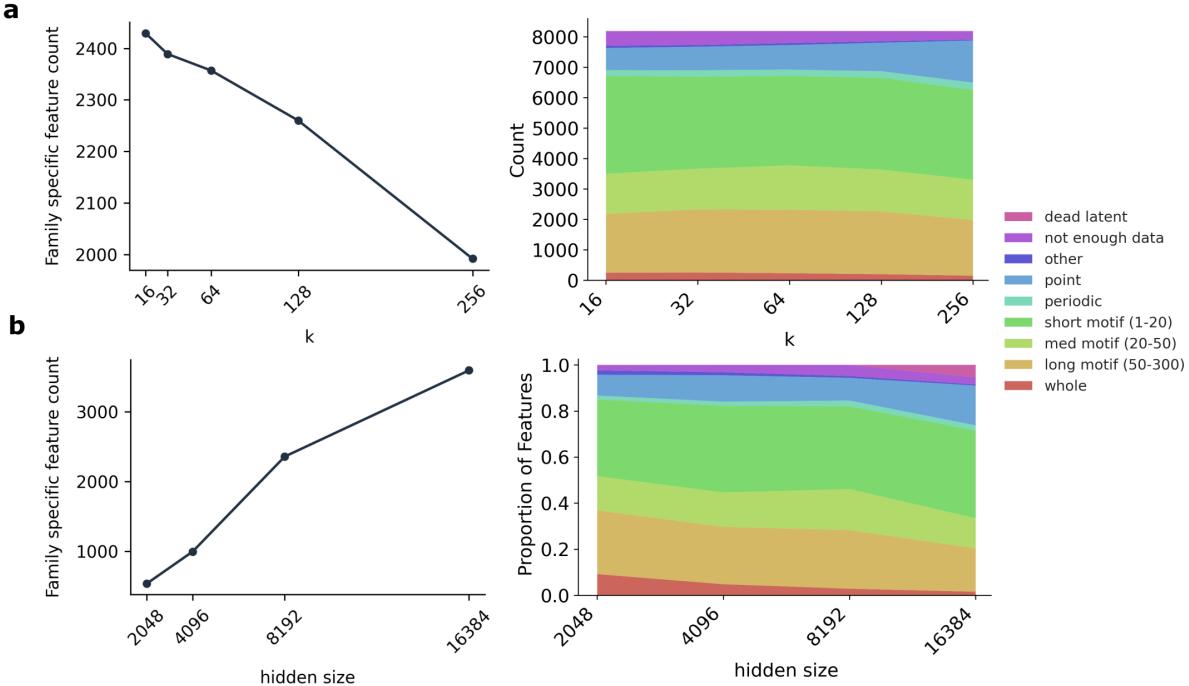
这种分类法 (表 2) 使他们能够量化超参数如何影响特征发现。例如,他们发现增加稀疏性 (降低 \(k\)) 会迫使模型学习更多的家族特异性特征,这可能是因为家族身份是一种高效压缩蛋白质信息的方式。
5. 可解释探测: 连接潜在特征与功能
识别特征很有趣,但我们能用它们来预测生物学特性吗?这就是可解释探测 (Interpretable Probing) 的用武之地。
计算生物学中的标准做法是在 pLM 的输出上训练一个线性分类器 (“探测器”) ,以预测诸如亚细胞定位等属性。虽然准确,但这些探测器是不可解释的。你得到了一个预测结果,但你不知道为什么。
研究人员尝试了一种不同的方法: 在 SAE 潜在特征上训练线性探测器。
性能 vs. 可解释性
首先,他们必须确保使用 SAE 潜在特征不会破坏性能。
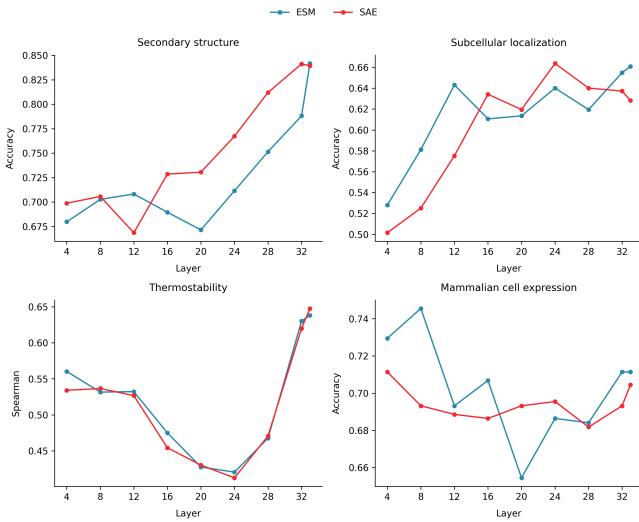
如图 5 所示,SAE 探测器 (红线) 在二级结构、亚细胞定位和热稳定性等任务上的表现与标准 ESM 探测器 (蓝线) 相当。在二级结构预测中,SAE 探测器实际上在许多层中都优于基线。
但真正的价值不在于准确性,而在于可解释性 。 由于探测器是线性的,研究人员可以查看具有最高正权重的潜在特征,以了解是什么生物学机制驱动了预测。
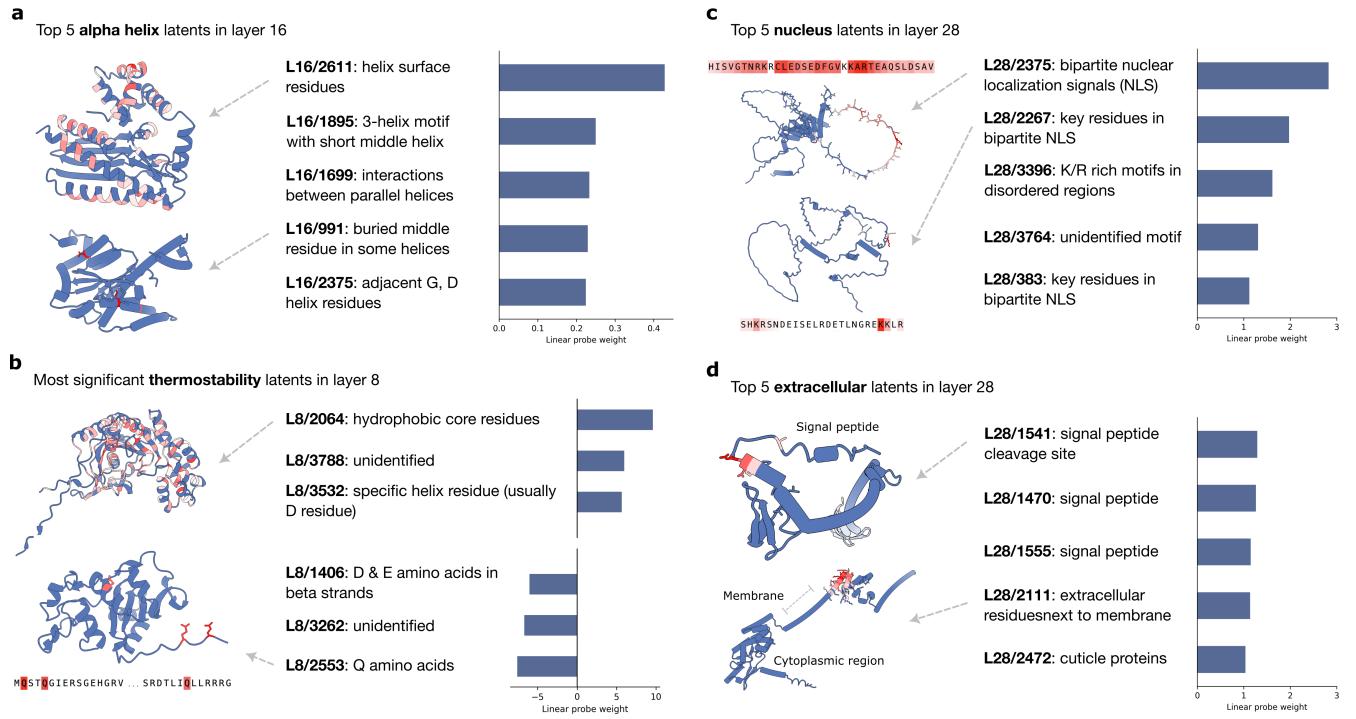
案例研究 1: 二级结构
在预测 α-螺旋时,探测器为那些明确检测螺旋结构的潜在特征分配了高权重。
同样,对于 β-折叠,探测器发现了追踪 β-链典型交替残基模式的潜在特征。
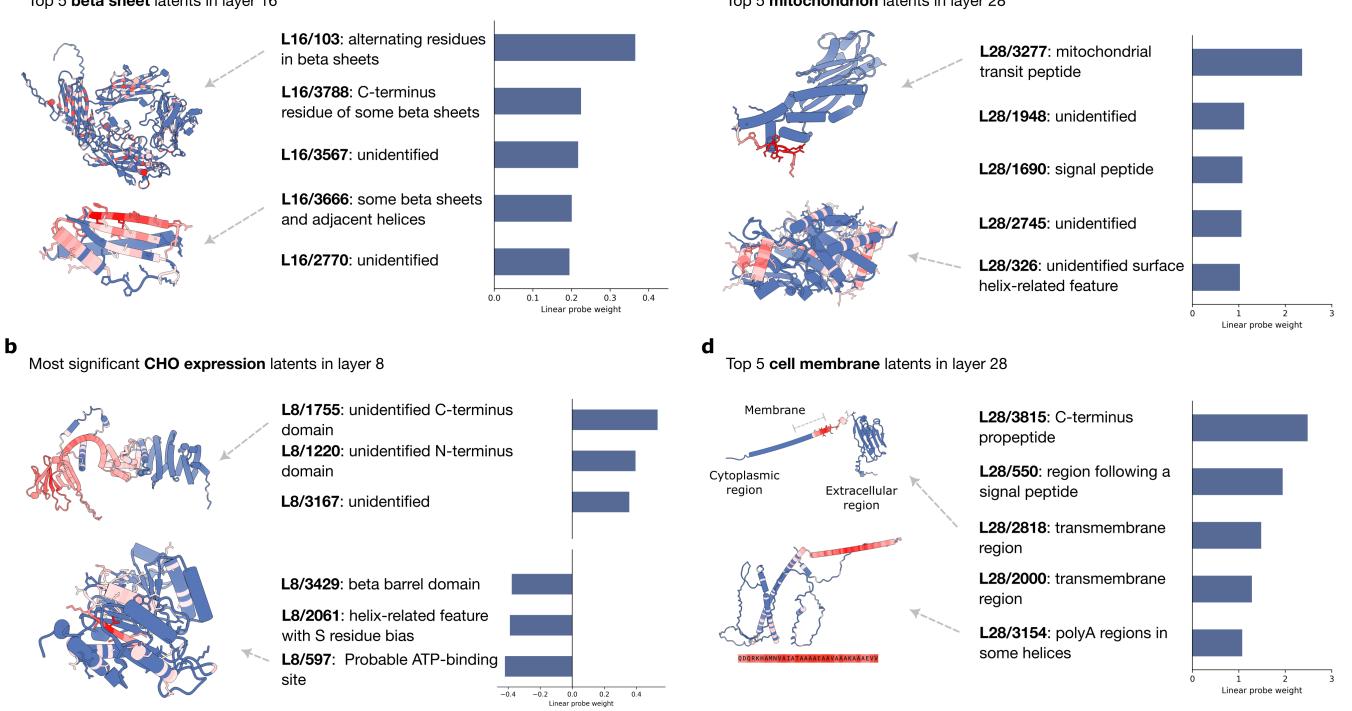
这证实了 SAE 已经学会了蛋白质折叠的“语法”。
案例研究 2: 亚细胞定位
这项任务涉及预测蛋白质在细胞中的去向 (例如,细胞核、线粒体、细胞膜) 。其生物学机制通常由“信号肽”驱动——即充当运输标签的短序列。
SAE 探测器在无监督的情况下重新发现了这些生物学机制。
- 细胞核: 顶部特征 (图 6c) 检测核定位信号 (NLS) , 特别是识别由连接子分隔的精氨酸 (R) 和赖氨酸 (K) 残基模式。
- 细胞外/膜: 模型发现了对应于信号肽和跨膜区域的特征 (图 9d) 。
(注: 图 9 指的是上文显示的线粒体/膜面板) 。
这是一个强有力的概念证明: SAE 识别出的正是生物学家花费数十年通过实验表征的序列模体。
案例研究 3: 热稳定性
预测蛋白质的耐热程度被证明是一个不同的挑战。先前的研究表明,pLM 在这项任务上难以扩展。
SAE 分析 (图 6b) 揭示了原因。模型并没有学习关于蛋白质稳定性的复杂生物物理学。相反,最具预测性的潜在特征本质上是在计算氨基酸。
- 正权重: 疏水核心残基 (可稳定蛋白质) 。
- 负权重: 谷氨酰胺 (Q),与不稳定性相关。
SAE 的可解释性暴露了模型的策略: 对于这个特定任务,它依赖于简单的组成统计数据,而不是深刻的结构理解。
案例研究 4: 哺乳动物细胞表达
研究人员还测试了一项实际的工业任务: 预测人类蛋白质是否可以在 CHO (中国仓鼠卵巢) 细胞中成功表达,这对于药物制造至关重要。
SAE 发现了一个与表达呈负相关的潜在特征,该特征检测ATP 结合位点 (图 9b) 。这产生了一个合理的生物学假设: 也许表达这种蛋白质会通过隔离 ATP 干扰宿主细胞的代谢,导致表达失败。这说明了 SAE 如何从解释转向假设生成。
为什么 SAE 探测器在解释方面更好
SAE 探测器中的权重分布与标准探测器有着根本的不同。
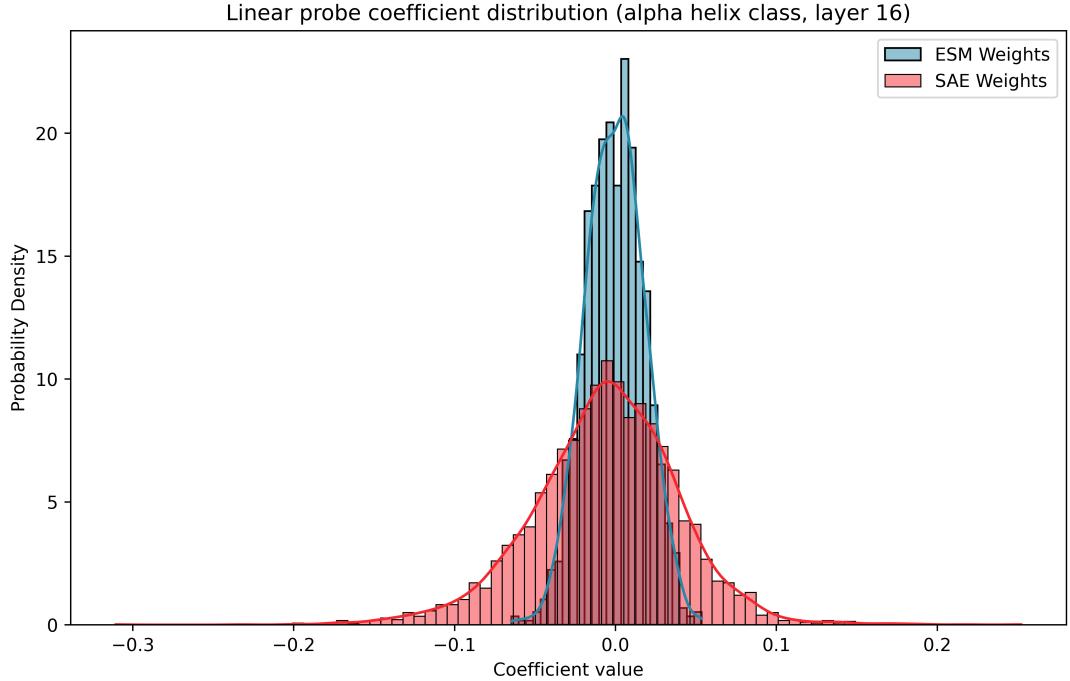
如图 10 所示,SAE 系数 (红色) 具有更高的方差和不同的偏度。这表明 SAE 潜在特征更加“单义”——每一个都是独特的预测因子——而 ESM 权重是许多微小贡献的混乱组合。
6. 通过操控验证特征
研究人员更进一步,验证了他们的“家族特异性”假设。他们进行了“操控”实验,即人工钳制某个特征的激活值,并运行模型以生成序列。
他们比较了操控家族特异性潜在特征与随机潜在特征的效果。
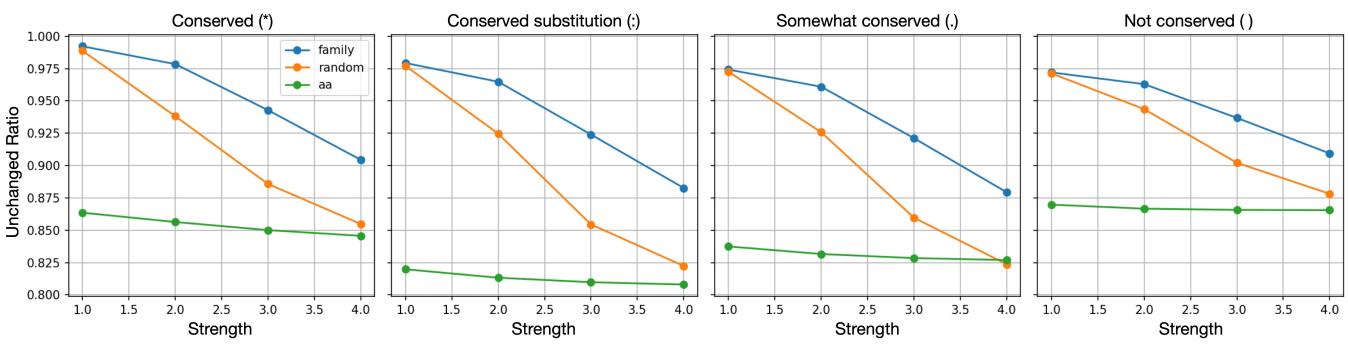
图 7 显示了残基的“未变比例”。当他们操控家族特异性潜在特征 (蓝线) 时,序列的变化小于操控随机潜在特征时的变化,特别是对于保守残基。这表明家族特异性潜在特征已深深融入模型对该蛋白质进化约束的理解中。
7. 结论: 迈向机制生物学
从“机械可解释性” (理解模型) 到“机制生物学” (理解生命) 的转变是这项技术的终极承诺。
Adams、Bai 及其同事已经证明,稀疏自编码器不仅仅是调试 AI 的工具。它们是观察 pLM 学习到的高维数据的显微镜。通过将蛋白质表征分解为可解释的特征,我们可以:
- 验证知识: 确认模型使用已知的生物学知识 (如 NLS 模体) 进行预测。
- 审计可靠性: 检测模型何时依赖启发式方法 (如用于热稳定性的氨基酸计数) 而非物理原理。
- 生成假设: 为理解不足的任务 (如 CHO 表达) 识别预测特征,以指导实验生物学。
蛋白质语言模型的“黑盒”正在开始打开,揭示出内部一个结构化的、可学习的生物学字典。
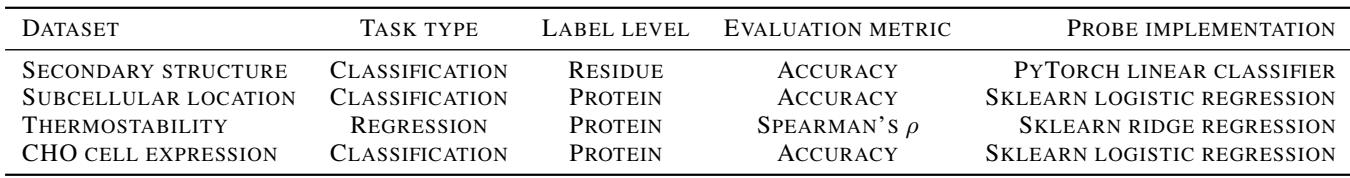
下游任务摘要: