](https://deep-paper.org/en/paper/2504.11454/images/cover.png)
构建更佳的蛋白质模型: 如何修复多模态 AI 中的“结构鸿沟”
蛋白质是生命的分子机器。为了理解生物学——并设计新药——我们需要理解蛋白质的两种不同“语言”: 它们的序列 (氨基酸串) 和它们的结构 (它们如何折叠成 3D 形状) 。
历史上,AI 将这两者视为独立的问题。我们有像 ESM 这样用于读取序列的模型,以及像 AlphaFold 这样用于预测结构的模型。但最近,研究人员一直试图将它们融合为多模态蛋白质语言模型 (Multimodal Protein Language Models, PLMs) 。 理想情况下,一个单一的模型应该能够读取序列,理解其几何结构,并生成既符合化学原理又在结构上合理的全新蛋白质。
然而,这其中存在一个陷阱。当前的多模态模型难以捕捉 3D 结构的细粒度细节。它们通常依赖于将 3D 坐标“词元化 (tokenizing) ”为离散符号,这一过程会丢失信息。
在最近的一篇论文《Elucidating the Design Space of Multimodal Protein Language Models》 (阐明多模态蛋白质语言模型的设计空间) 中,研究人员系统地拆解了这个问题。他们找出了当前模型失败的原因,并提出了一套涵盖生成式建模、架构设计和数据策略的解决方案——这些方案显著提升了模型性能。
在这篇文章中,我们将剖析他们的发现,并解释他们是如何设法让一个 6.5 亿 (650M) 参数的模型在蛋白质折叠任务中击败 30 亿 (3B) 参数的基线模型的。
问题所在: 当词元化失效时
为了将 3D 蛋白质结构输入到语言模型 (它期望的是类似文本的输入) 中,我们通常使用结构词元化器 (Structure Tokenizer) 。 它将连续的 3D 坐标 (\(x, y, z\)) 转换成离散的整数序列 (词元/tokens) ,就像 ChatGPT 处理单词的方式一样。
研究人员分析了 DPLM-2 (一种最先进的多模态模型) ,并确定了该过程中存在的三个主要瓶颈:
- 信息丢失 (Information Loss) : 将连续坐标转换为离散词元就像是一种有损压缩算法。你会丢失精细的细节。
- “重构”陷阱 (The “Reconstruction” Trap) : 一个词元化器可能擅长压缩和解压结构 (重构) ,但这并不意味着语言模型能有效地生成这些词元。
- 索引预测难题 (The Index Prediction Problem) : 预测词元的精确索引 (例如“Token #4532”) 非常困难。模型经常会搞错具体的索引,即使它理解的结构概念是接近的。
如下表所示,虽然模型很难预测准确的索引 (准确率仅约 8-12%) ,但它实际上能更好地预测结构底层的位 (二进制表示) (准确率约 77-86%) 。
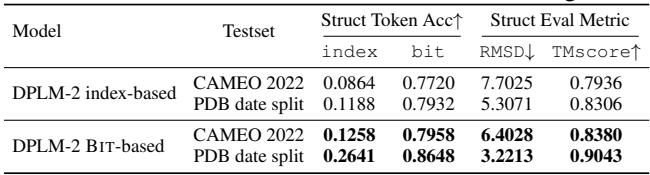
这一观察结果——即模型对结构的“认知”比离散索引所表现出的要好得多——构成了他们第一个重大改进的基础。
解决方案 1: 改进的生成式建模
研究人员提出了一个新的设计空间来修复这些生成问题,总结在下图中。
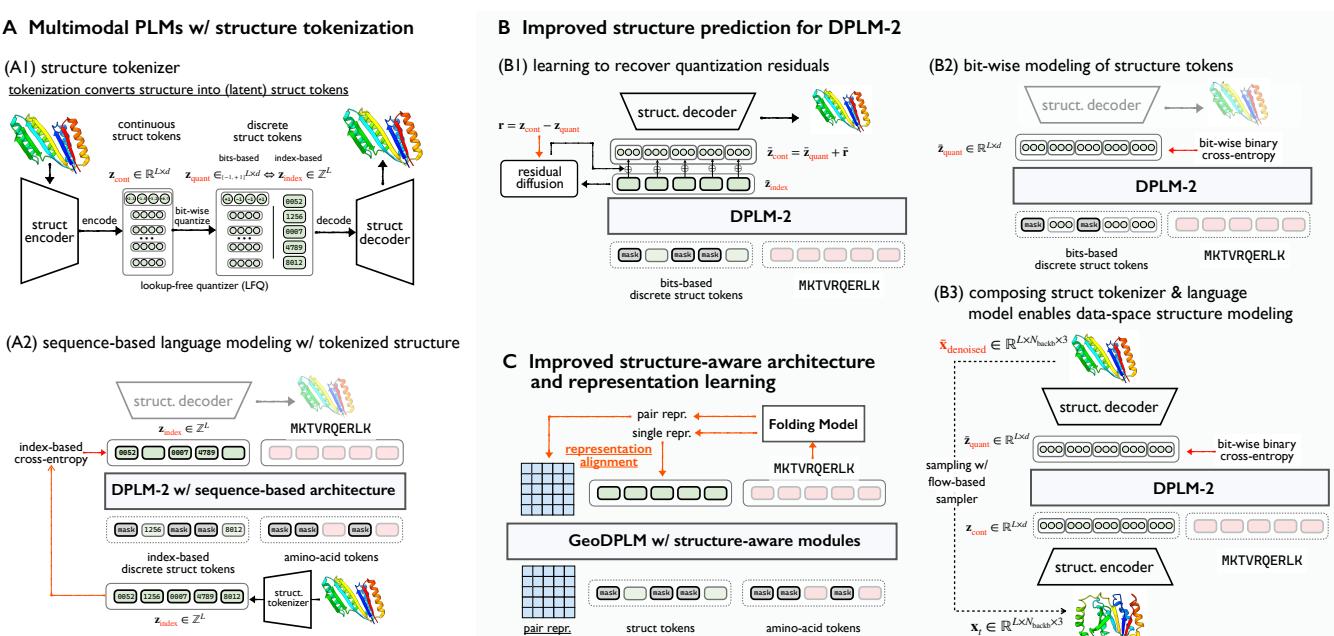
按位离散建模 (Bit-wise Discrete Modeling)
由于从成千上万个选项中预测一个特定的整数非常困难 (且容易出错) ,研究人员改变了监督目标。他们不再训练模型预测词元索引,而是预测构成该词元的位 (bits) 。
这利用了词元化器中使用的“无查找量化” (Lookup-Free Quantization, LFQ) 方法。如果一个词元由二进制代码表示 (例如 10110) ,那么独立预测每一位是一个比从 262,144 个选项的字典中挑选一个数字要简单得多的分类任务。这创造了一个更容易让模型学习的“细粒度”监督信号。
利用 RESDIFF 恢复丢失细节
即使有完美的词元预测,离散词元仍然是 3D 结构的“有损”压缩。为了解决这个问题,团队引入了一个轻量级的残差扩散 (Residual Diffusion, ResDiff) 模块。
可以将离散词元想象成蛋白质的“低分辨率”支架。ResDiff 模块充当抛光器的角色;它学习“残差”——即块状的词元化结构与平滑的真实连续结构之间的差异。
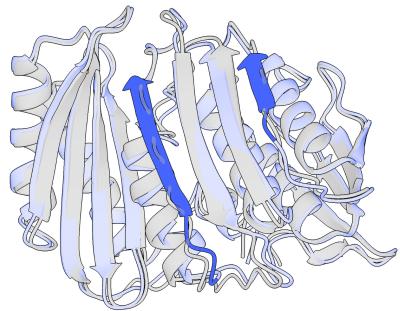
这种效果在视觉上非常显著。在下图中,你可以看到残差扩散 (蓝色) 如何优化初始预测 (灰色) ,修复断裂的环 (loops) 并更紧密地对齐二级结构。
解决方案 2: 几何架构 (GeoDPLM)
标准的语言模型是为 1D 序列 (文本) 设计的。然而,蛋白质本质上是受物理和空间关系支配的几何物体。将蛋白质纯粹视为 1D 词元串忽略了这一现实。
为了解决这个问题,研究人员引入了 GeoDPLM , 将“几何感知”模块集成到架构中。
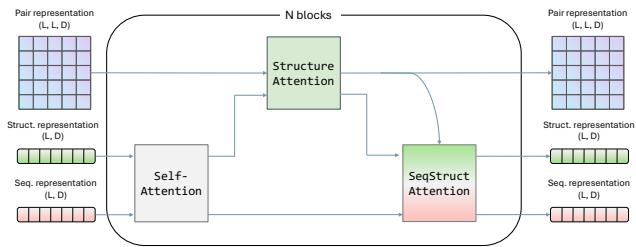
受 AlphaFold 的启发,GeoDPLM 在蛋白质语言模型 (PLM) 中引入了两个关键概念:
- 成对表示 (Pair Representations) : 模型不仅关注单个残基,还显式计算和更新成对残基的信息 (即残基 \(i\) 与残基 \(j\) 的关系) 。
- 三角更新 (Triangle Updates) : 这些是专门的数学运算,用于强制几何一致性 (例如,如果点 A 靠近 B,且 B 靠近 C,那么 A 必须相对靠近 C) 。
研究人员添加了一个结构注意力模块 (Structure Attention Module) (如下图所示) ,它将这些成对表示与标准的序列信息一起处理。
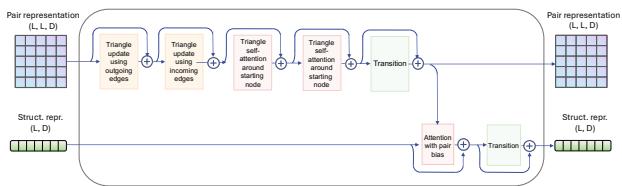
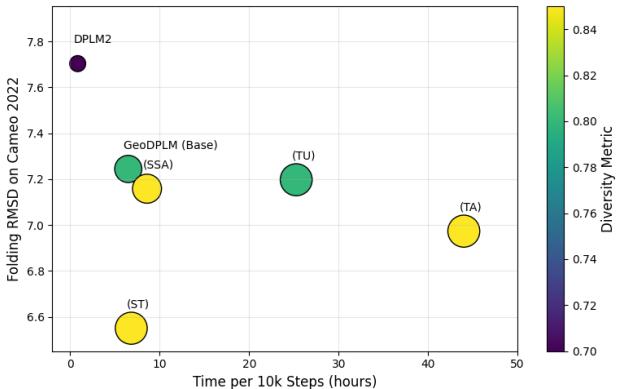
权衡: 虽然这些几何模块显著提高了折叠精度 (降低了 RMSD) ,但它们的计算成本很高。例如,“三角注意力”机制使训练速度减慢了近 7 倍。研究人员找到了一个最佳平衡点 (GeoDPLM Base) ,它使用转换层和成对偏差来获得大部分收益,而没有巨大的速度惩罚。
解决方案 3: 表示对齐 (REPA)
改进的第三大支柱是表示对齐 (Representation Alignment, REPA) 。
这个想法很简单: 我们已经拥有专门的“折叠模型” (如 ESMFold) ,它们非常擅长理解结构。为什么不把它们的知识蒸馏到我们的多模态模型中呢?
研究人员不仅在离散词元 (这是“尖锐”且严苛的目标) 上训练模型,还强制 PLM 的内部表示与 ESMFold 的丰富、连续表示相对齐。这提供了一个更平滑、高维的学习信号。
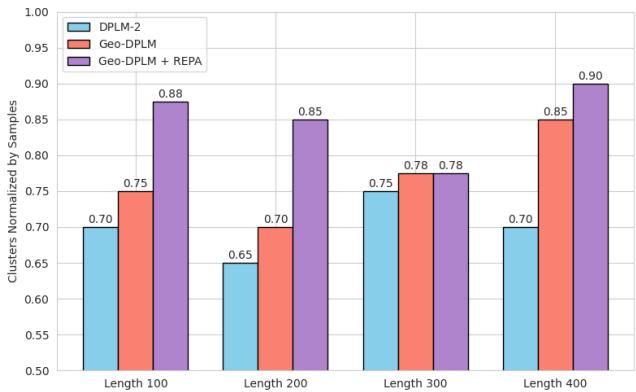
这为什么重要? 它极大地改善了多样性 。 多模态 PLM 经常遭受“模式崩塌 (mode collapse) ”的困扰,即它们反复生成非常相似的结构。如下图所示,与基础模型相比,添加 REPA (紫色柱) 显著增加了生成蛋白质的多样性。
数据的作用: 单体 vs. 多聚体
大多数蛋白质语言模型仅在单体 (monomers) (单条蛋白质链) 上进行训练。但生物学是交互式的;蛋白质会形成称为多聚体 (multimers) 的复合物。
研究人员整理了一个名为 PDB-Multimer 的数据集,并发现了一个有趣的关系:
- 在多聚体数据上进行训练有助于模型理解链之间的相互作用。
- 至关重要的是, 在多聚体上训练可以提高单体的性能。
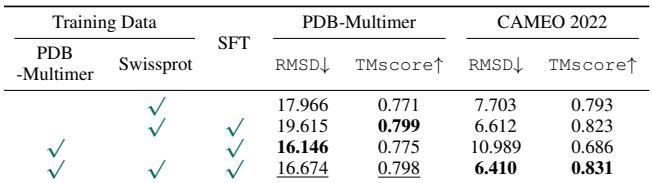
这表明蛋白质结构的“语法”是通用的。学习两条独立的链如何相互作用,有助于模型理解单条链如何自行折叠。下表显示,使用多聚体数据进行微调可以改善复杂情况下的重构和折叠指标。
融会贯通: 结果展示
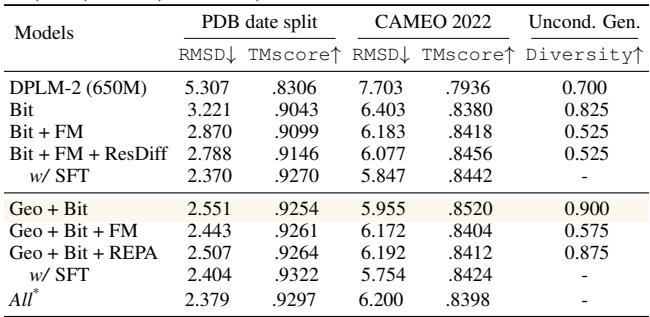
那么,当你结合按位建模、几何架构和表示对齐时会发生什么呢?
研究人员在标准的“蛋白质折叠”任务 (从序列预测结构) 上测试了他们的方法。使用的指标是 RMSD (均方根偏差) ——数值越低越好。
- 基线 (DPLM-2): 5.52 Å
- 使用按位建模: 3.22 Å (巨大的飞跃!)
- 使用几何架构 + 按位建模 + SFT (有监督微调): 2.36 Å
值得注意的是,在 PDB 数据集上,他们的 6.5 亿 (650M) 参数模型击败了 30 亿 (3B) 参数的基线 (ESMFold 约为 3B) 。
下表总结了这些方法的正交性。“推荐设置” (Geo + Bit) 在性能和效率之间提供了最佳平衡。
结论
这项研究强调,对于多模态蛋白质生成而言,简单地向标准语言模型投喂更多数据是不够的。 监督信号的设计 (位 vs. 索引) 和架构的归纳偏置 (几何模块) 起着巨大的作用。
通过“阐明这一设计空间”,作者们为下一代蛋白质 AI 提供了蓝图。这些模型不仅仅是预测静态结构;它们正在学习蛋白质形式和功能的统一表示,为更精确的*从头 (de novo) *蛋白质设计铺平道路。
关键要点:
- 不要预测索引;预测位。 这是教 PLM 学习结构的更有效方法。
- 几何很重要。 你不能将 3D 结构严格视为 1D 文本;你需要几何模块。
- 蒸馏有帮助。 与专门的折叠模型对齐可以提高生成的多样性。
- 多聚体很重要。 来自蛋白质复合物的数据有助于模型更好地理解单个蛋白质。