](https://deep-paper.org/en/paper/4192_p_all_atom_is_unlocking_n-1776/images/cover.png)
蛋白质设计长期以来一直被描述为“逆蛋白质折叠问题”。如果说折叠是大自然将氨基酸序列转化为 3D 结构的方式,那么设计就是我们要寻找能够折叠成特定、期望形状的序列的尝试。
多年来,这一领域一直被“分而治之”的策略所主导。研究人员通常先生成主链结构 (即丝带状结构) ,然后使用单独的模型将序列和侧链“填充”到该主链上。虽然这种方法有效,但它忽略了一个基本的生物学现实: 蛋白质的主链与其侧链是紧密相连的。原子的特定化学性质决定了折叠方式,而折叠方式又决定了哪些原子适合放置其中。
Pallatom 应运而生,这是一种旨在弥合这一鸿沟的新型生成模型。通过同时对结构和序列的联合分布进行建模——特别是通过关注所有原子的概率 (\(P(all\text{-}atom)\)) ——Pallatom 在生成高保真、多样化和新颖的蛋白质方面取得了最先进的成果。
在这篇文章中,我们将解构 Pallatom 论文。我们将探讨解决了可变原子问题的巧妙的“atom14”表示法、驱动模型的双轨架构,以及表明我们正进入协同生成蛋白质设计新时代的实验结果。
核心问题: 序列与结构的脱节
要理解 Pallatom 的重要性,我们需要看看蛋白质建模的现状。历史上,该领域依赖于两个条件概率:
- \(P(structure \mid seq)\): 从已知序列预测结构 (例如 AlphaFold) 。
- \(P(seq \mid backbone)\): 为固定主链设计序列 (例如 ProteinMPNN) 。
大多数生成流程将这些步骤串联起来。它们可能首先通过幻觉生成一个主链结构,然后为其优化序列。在数学上,这近似于将联合概率表示为 \(P(backbone) \cdot P(seq \mid backbone)\)。
局限性在于?这种分步过程将主链与侧链解耦了。它假设你可以在不知道氨基酸组成的情况下设计出完美的主链。然而,在自然界中,庞大的侧链、氢键和疏水相互作用往往决定了主链的扭转和转向。通过在初始生成阶段忽略这些全原子相互作用,我们限制了设计空间和准确性。
Pallatom 旨在直接对联合分布 \(P(structure, seq)\) 进行建模。它将蛋白质视为一团原子云,其中几何形状本身就编码了序列信息。
“atom14”表示法: 解决可变原子难题
全原子生成的第一个障碍是表示法。在扩散模型中,我们通常将噪声转化为数据。但是,如果我们还不知道氨基酸序列,我们就不知道每个残基中有多少个原子。甘氨酸 (Glycine) 只有 4 个重原子;色氨酸 (Tryptophan) 则有 14 个。如何为一个粒子数量未知且不断变化的系统构建神经网络?
Pallatom 引入了 atom14 , 这是一个统一的表示框架。
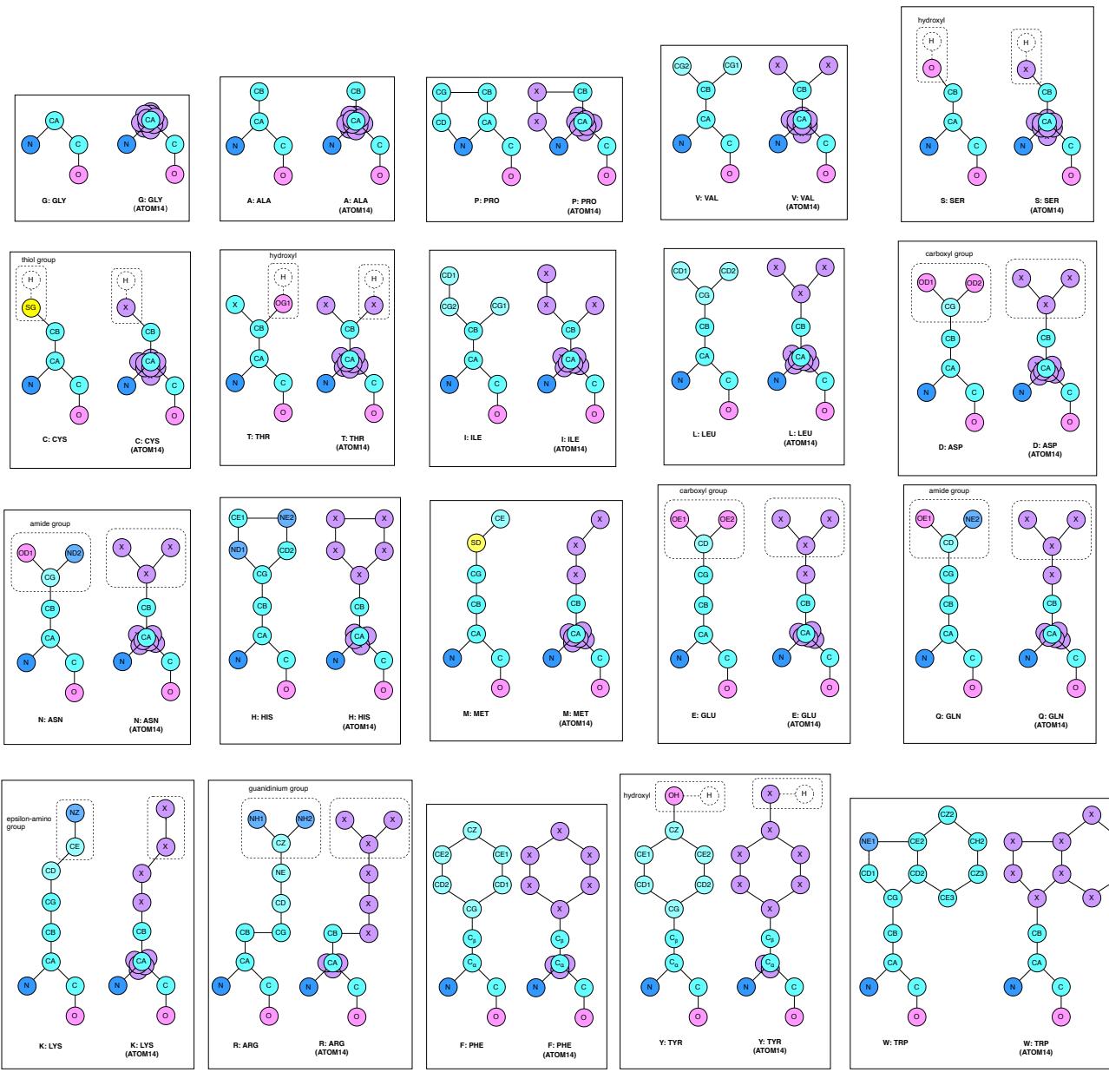
如 图 3 所示,atom14 将每个残基标准化为 14 个原子的固定大小。这里有一个巧妙之处:
- 标准化: 蛋白质中的每个位置都被分配了最多容纳 14 个原子的插槽。
- 虚拟原子: 对于较小的氨基酸,“多余的”原子插槽并不是空的 (如果是空的就需要知道类型) ,而是被折叠到 \(\alpha\) 碳原子 (\(C_\alpha\)) 的坐标上。
- 无泄露: 在生成开始时,模型不需要知道侧链原子的具体元素。它将它们视为点云中的通用“被掩码”粒子。
这使得模型能够幻觉生成可能侧链的“叠加态”。随着结构在扩散过程中逐渐精细化,这 14 个点的几何形状开始类似于某种特定的氨基酸。这消除了设计序列和结构之间的冲突——它们从噪声中共同涌现。
Pallatom 架构
Pallatom 采用基于扩散的框架。具体来说,它使用了 EDM (Elucidating the Design Space of Diffusion-Based Generative Models) , 这是一种用于生成坐标数据的稳健公式。
扩散过程
目标是逆转一个过程,该过程逐渐向蛋白质结构添加噪声,直到它变成一团随机的点云。生成过程 (去噪) 由微分方程定义:
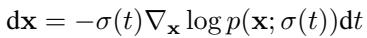
模型学习一个函数 \(D_\theta\),该函数从噪声输入中预测干净、去噪的坐标。这是使用加权均方误差损失进行训练的:
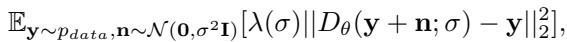
在实践中,网络在每一步都预测最终的干净结构,扩散调度器在预测结果和当前噪声状态之间进行插值,以此向前推进一步。
主干网络 (MainTrunk) 和原子解码器 (AtomDecoder)
驱动这一过程的神经网络非常复杂。它需要同时处理两个层面的信息: 残基层面 (氨基酸序列) 和原子层面 (3D 点云) 。
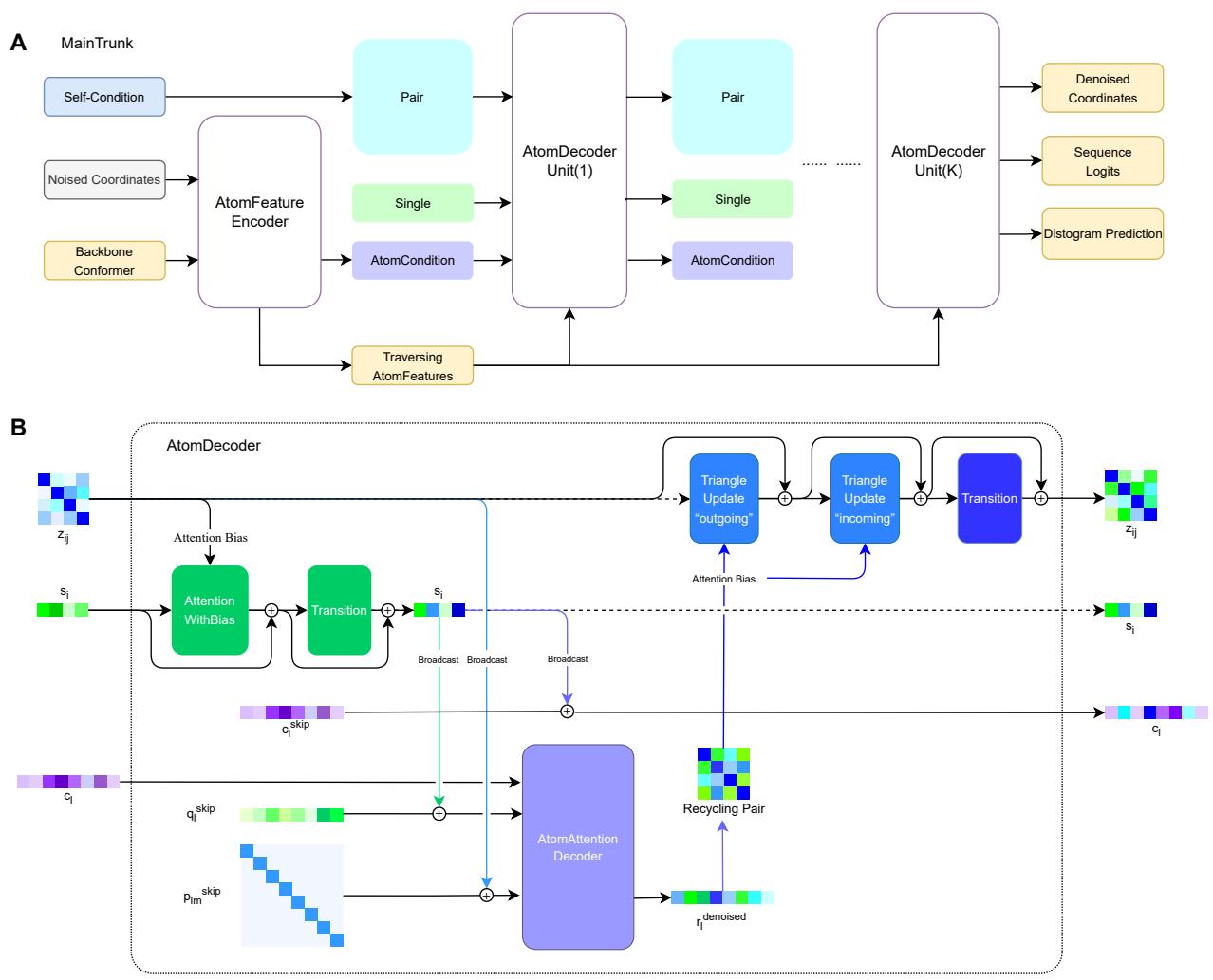
图 1 展示了该架构,它由两个主要部分组成:
- MainTrunk (图 1A) : 这是编码器。它接收噪声坐标和“自条件化”信息 (来自上一步的猜测) 并初始化特征。它建立了一个 双轨表示 (Dual-Track Representation) :
- 残基轨道: 处理诸如相对位置和氨基酸之间配对相互作用的特征 (\(z_{ij}\) 和 \(s_i\)) 。
- 原子轨道: 处理每个残基 14 个原子的显式 3D 坐标。
- AtomDecoder (图 1B) : 这是模型的引擎。它包含多个解码单元,用于迭代地优化结构。
- 信息流: 它使用“遍历”表示法。信息从残基层面流向原子 (广播) ,再从原子流回残基。
- 三角更新: 借鉴 AlphaFold,它使用三角注意力机制来确保残基之间的成对距离在几何上是一致的。
- 循环 (Recycling): 关键在于,一个块的输出不仅仅传递给下一个块;预测的几何结构被“循环”回配对表示 (\(z_{ij}\)) 中,使模型能够随时间推移优化其对全局拓扑的理解。
第 \(k\) 步的坐标更新规则结合了输入与网络的预测更新,并按噪声因子缩放:
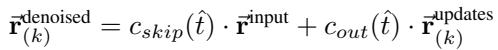
从几何到序列
Pallatom 生成的是 3D 结构,但我们最终需要的是字母序列 (氨基酸) 。由于 atom14 表示法如此精确地编码了侧链几何形状,因此序列实际上是隐式包含在结构中的。
如果模型生成了一个完美的双环侧链形状,那它必须是色氨酸。如果它生成了一个短的分支链,那就是缬氨酸或苏氨酸。
Pallatom 使用一个名为 SeqHead 的模块。它获取原子的最终几何嵌入,将它们聚合,并通过一个简单的线性层来预测 20 种氨基酸中每一种的概率。这是一种在生成结束时自然发生的“结构到序列”的解码。
训练目标
为了训练 Pallatom,研究人员使用了复合损失函数,以强制实现局部化学有效性和全局结构连贯性。
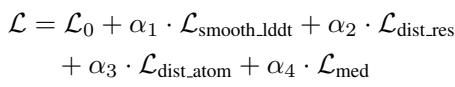
总损失 \(\mathcal{L}\) 包括:
- \(\mathcal{L}_{atom}\): 原子坐标上的标准扩散损失 (MSE)。
- \(\mathcal{L}_{seq}\): 交叉熵损失,确保预测的序列与真实值匹配。
- \(\mathcal{L}_{smooth\_lddt}\): 基于局部距离差异测试 (LDDT) 的损失。这优化了模型的局部几何形状 (如键长和侧链堆积) ,这对于物理真实性至关重要。
- \(\mathcal{L}_{dist}\): 距离图损失,监督残基和原子之间的成对距离,帮助模型学习全局拓扑。
实验结果
学习 \(P(all\text{-}atom)\) 真的比拆分任务效果更好吗?结果表明答案是肯定的。
可设计性和多样性
研究人员将 Pallatom 与其他最先进的模型进行了基准测试,包括 Protpardelle (另一种全原子模型) 、ProteinGenerator、Multiflow 和 RFDiffusion (仅主链) 。
关键指标是 DES-aa (全原子可设计性) : 如果我们取出生成的序列并使用 ESMFold 将其折叠,它是否与生成的结构匹配?
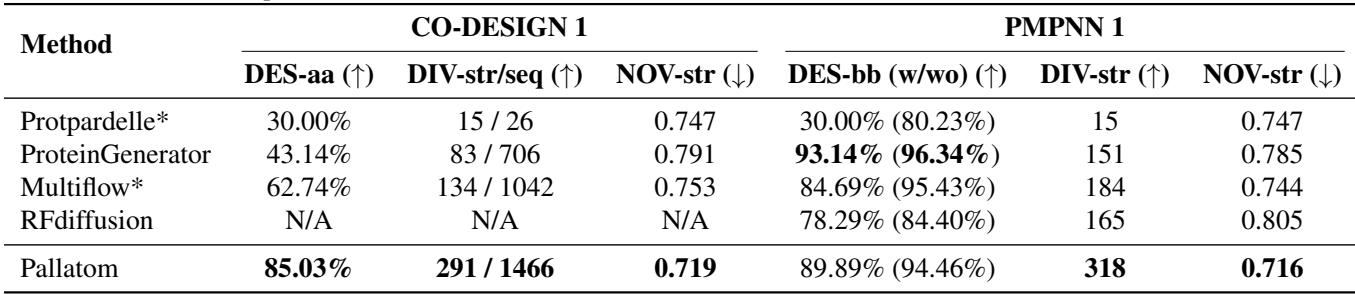
表 1 突显了性能差距。Pallatom 在 DES-aa 中达到了惊人的 85.03% , 显着优于 Protpardelle (30%) 和 Multiflow (62.74%)。
更令人印象深刻的是 多样性 (DIV-str) 。 生成模型通常遭受“模式坍塌”的困扰,即反复生成相同的几种安全结构。Pallatom 生成了 291 个独特的结构簇,几乎是 RFDiffusion (165) 的两倍,并且远超 ProteinGenerator (83)。
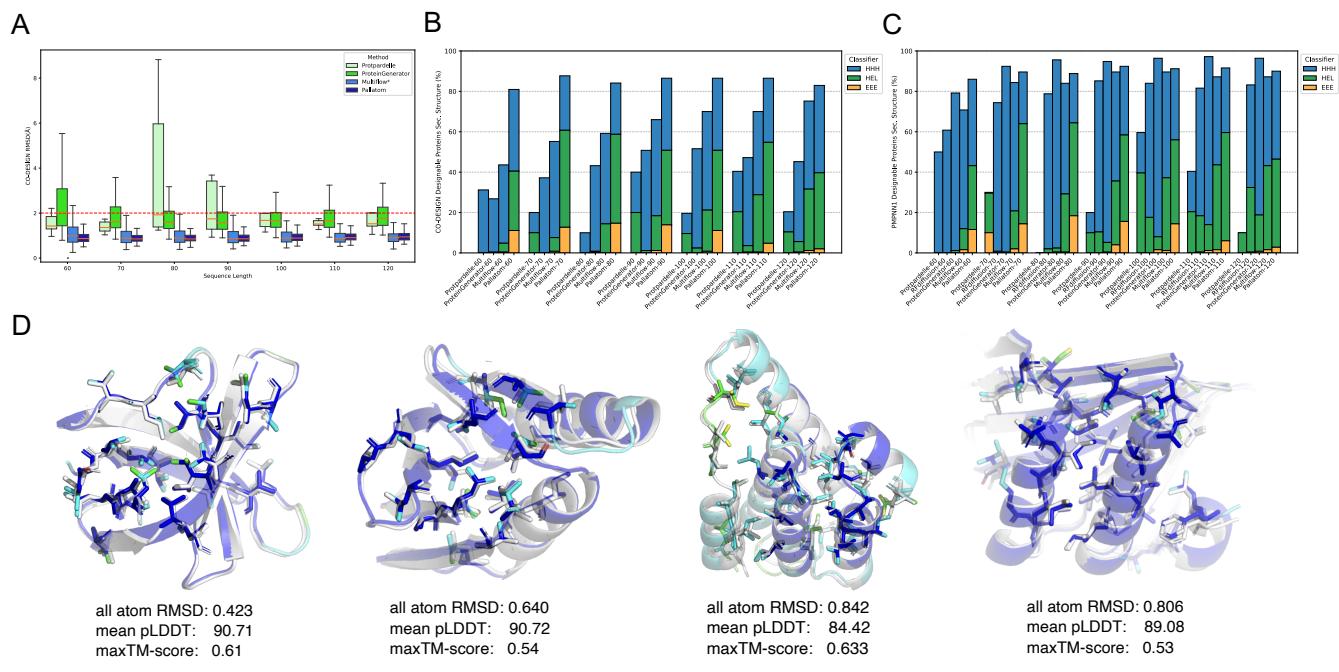
图 2 提供了视觉确认。图板 A 显示生成结构与折叠结构之间的 RMSD (误差) 始终很低。图板 D 展示了生成的蛋白质: 它们结构紧凑、折叠良好,并展现出清晰的二级结构。
扩展到更大的蛋白质
生成模型面临的最困难测试之一是 分布外 (OOD) 泛化。在一个较小蛋白质上训练的模型能否生成大型、复杂的蛋白质?
研究人员在长达 400 个残基的蛋白质上测试了 Pallatom (这明显长于训练时的裁剪尺寸) 。
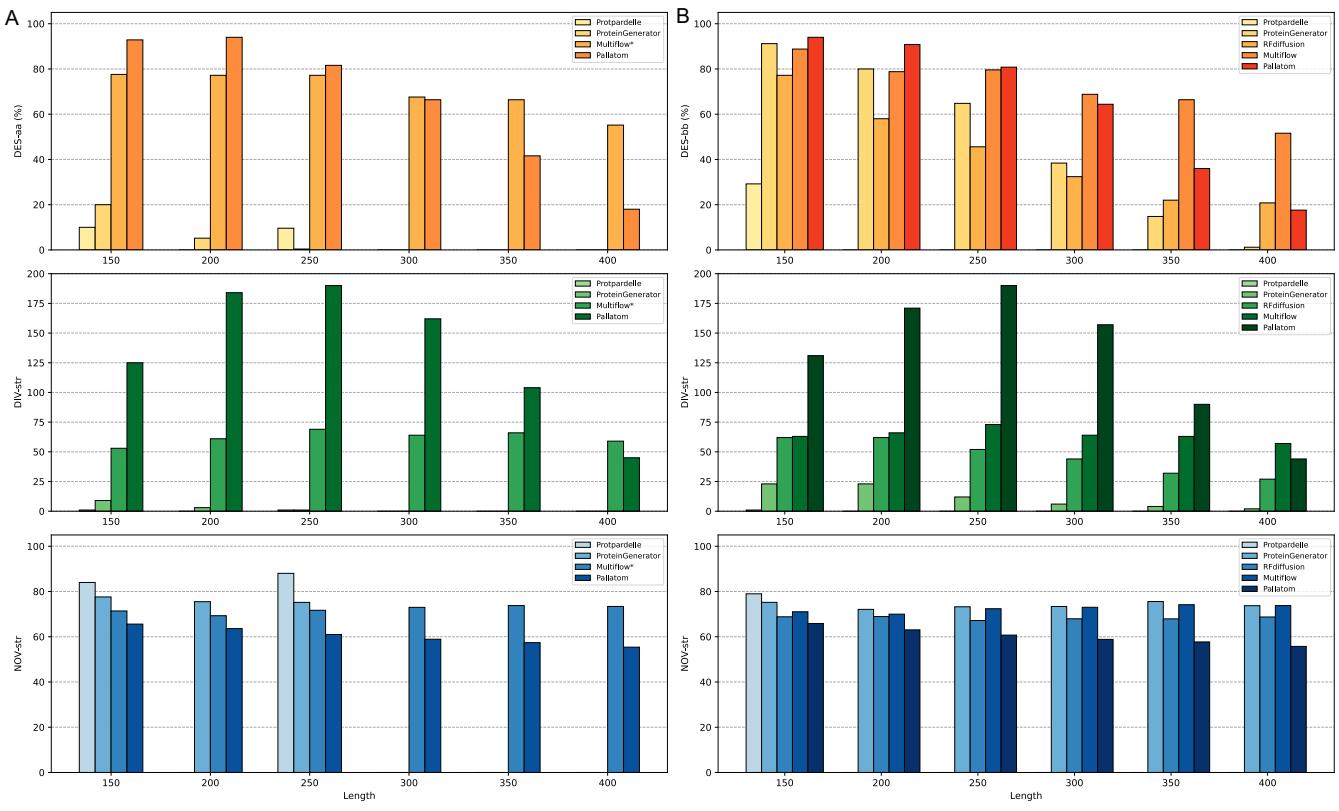
如 图 4 所示,Pallatom (紫色柱) 即使在长度 300 以上时仍保持较高的可设计性,而其他方法开始失效。它是唯一在长度 400 时仍能稳健生成全原子结构的方法。
物理真实性
蛋白质设计中的一个常见失败模式是“幻觉生成”出看起来像丝带一样漂亮,但在化学上不可能存在的结构 (例如,重叠的原子或拉伸的键) 。
Pallatom 对全原子坐标的关注在这里得到了回报。研究人员比较了生成的蛋白质与天然蛋白质 (PDB) 中的键长和键角分布。
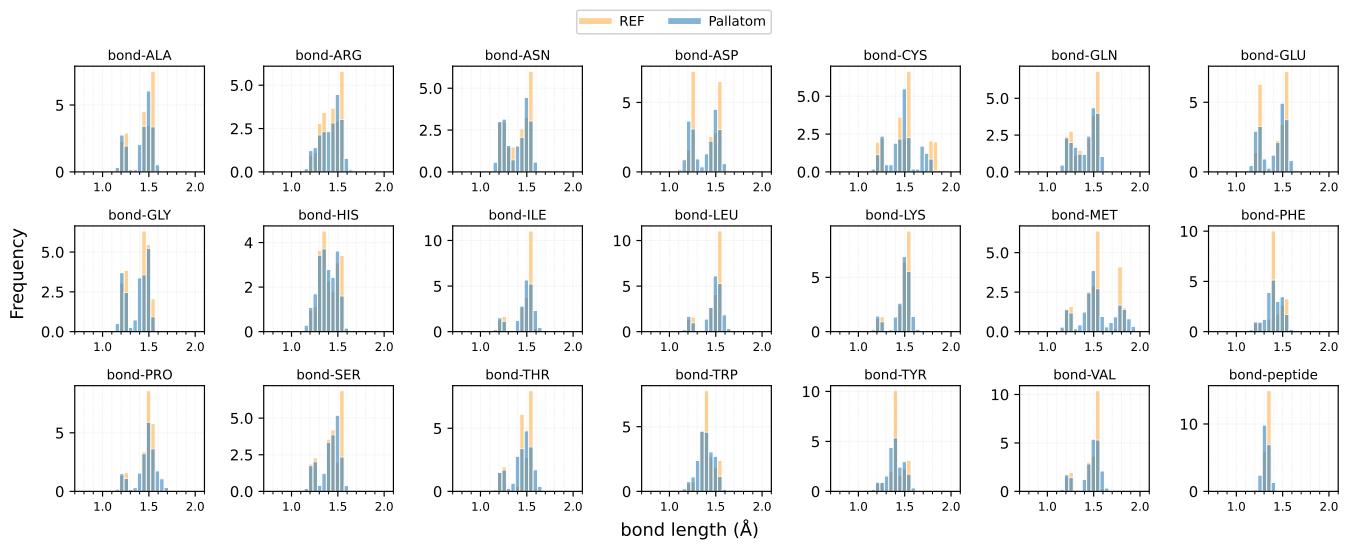
图 6 显示了键长分布。生成的蛋白质 (蓝色) 与天然参考 (黄色) 几乎完美吻合。
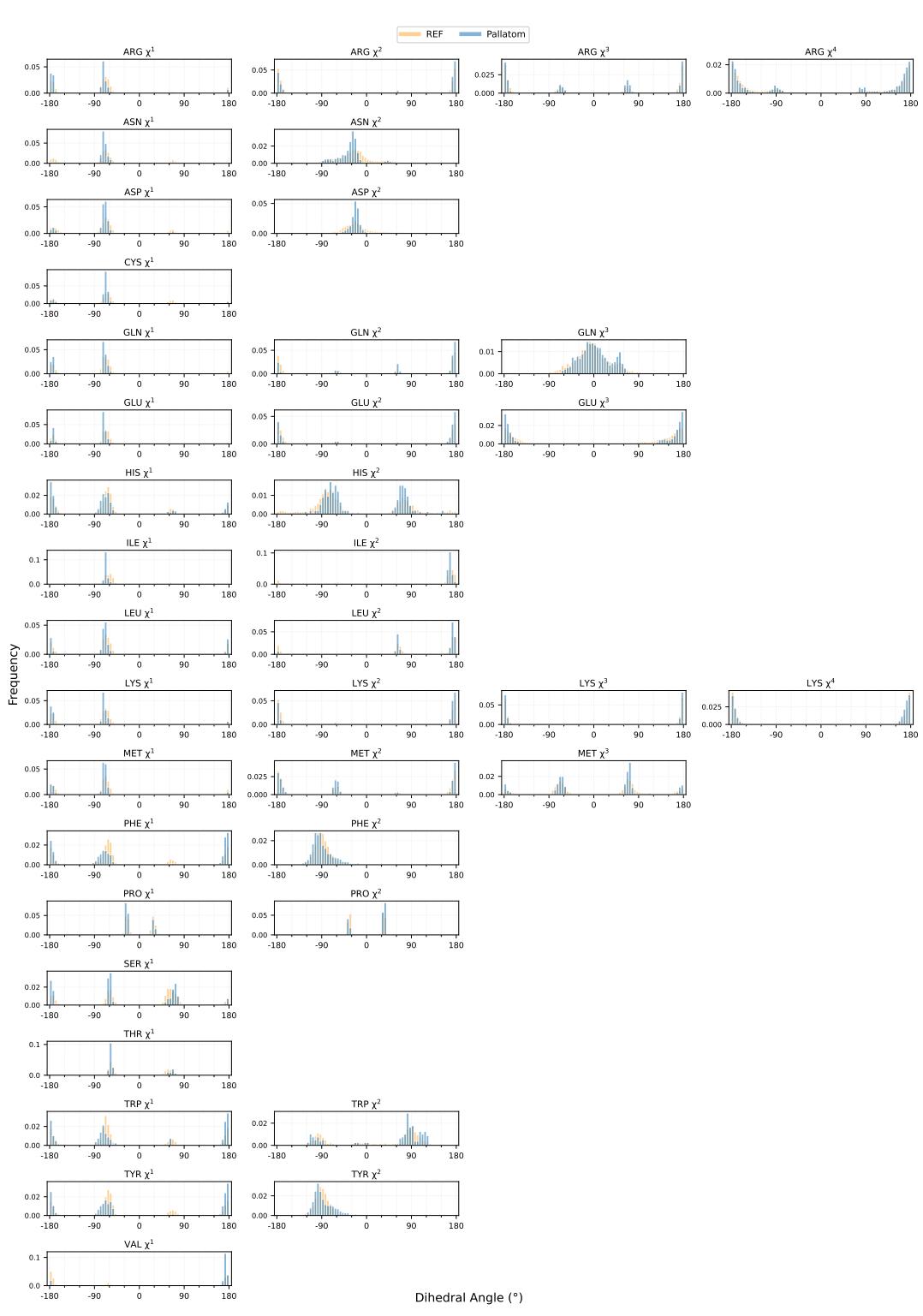
同样, 图 8 显示了 \(\chi\) 角 (侧链扭转角) 。这些角度决定了侧链如何堆积到蛋白质核心中。Pallatom 准确地重现了自然界中发现的旋转异构体状态,证明它已经学会了原子堆积的底层物理规律。
结论
Pallatom 代表了计算生物学向前迈出的重要一步。通过摒弃顺序的“先主链后序列”范式,并采用统一的 \(P(all\text{-}atom)\) 方法,它解决了定义蛋白质折叠的几何形状与化学性质之间的依赖关系。
引入 atom14 表示法和 双轨 架构使得模型能够以全原子细节进行“梦想”。结果是产生了一个不仅更准确,而且比其前身更多样化、更具可扩展性的生成器。
随着我们迈向设计复杂的酶、结合剂和分子机器,像 Pallatom 这样理解每一个原子之间复杂舞蹈的模型,将成为蛋白质工程师工具箱中必不可少的工具。